Senior Lecturer
School of Computer Science and Engineering, University of New South Wales
Biography
Sara Ballouz is a Senior Lecturer in Bioinformatics in the School of Computer Science and Engineering at the University of New South Wales.
Interests
- Transcriptomics (single-cell and bulk)
- Co-expression
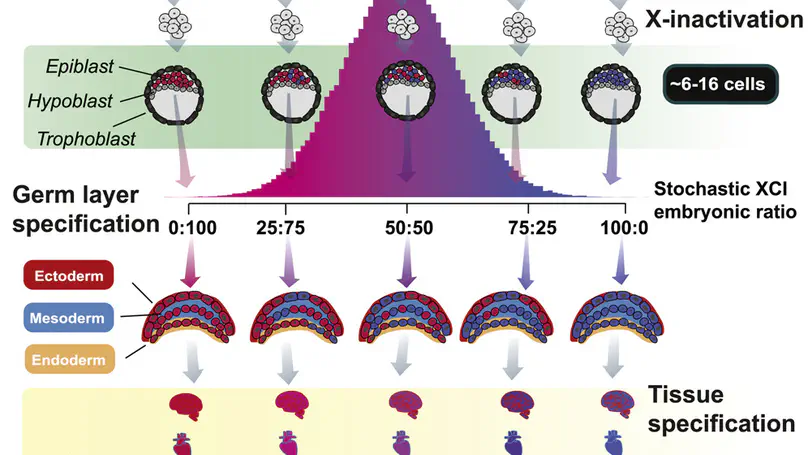
- X-linked disorders
- Sex differences in disease
- Meta-analysis
Education
PhD Bioinformatics, 2013
Victor Chang Cardiac Research Institute and University of New South Wales
BE Bioinformatics, 2008
University of New South Wales
BS Genetics, 2008
University of New South Wales